в московской школе
Выравнивание результатов секвенирования на геном вируса Ebola
|
Работа призёра конкурса проектов и исследований «Старт в медицину» открытой городской научно-практической конференции «Старт в медицину» в секции «Биотехнология и биоинженерия в медицине» |
Направление работы: Биоинформатика
Авторы работы: ГБОУ Школа № 2107
Email: Написать
Предметы: Биология, Информатика
Классы: 10 класс
Мероприятия: Конкурс проектов и исследований «Старт в медицину» открытой городской научно-практической конференции «Старт в медицину» 2021 года
|
Актуальность
В связи с обострением эпидемиологической ситуации в мире становится крайне актуальной задача быстрой идентификации природы патогенов. Современным быстрым и точным методом диагностики является секвенирование с выравниванием на референсный геном. Секвенирование ДНК или РНК человека c последующим выравниванием ридов на геном возбудителя болезни является точнейшим методом диагностики, позволяет в кратчайшие сроки установить причины заболевания и назначить необходимую процедуру лечения.
Цель
Анализ работы программ для выравнивания результатов секвенирования на геном вируса Ebola.
Задачи
1. Освоить работу с банками геномов и белков, банками результатов секвенирования.
2. Освоить геномный браузер IGV и интегрированный программный инструмент Ugene, программный пакет BLAST, программный пакет SRAtools.
3. Освоить биоинформатические инструменты Bowtie2, BWA, Minimap2, Hisat2, SAMtools и BAMtools.
4. Используя освоенные инструменты, выровнять результаты секвенирования на геном вируса Ebola. Провести сравнительный анализ их работы.
Оснащение и оборудование, использованное в работе
• Компьютер
• Операционная система Ubuntu
Описание
Для работы были взяты данные секвенирования, которые хранятся в формате SRA в виде парных прочтений, поэтому для распаковки использовался ключ --split-3 и выравнивались файлы как с прямыми прочтениями, так и с обратными.
При помощи программы fastq-dump был распакован SRA из пакета SRA Toolkit. В результате были получены два файла формата FASTQ с прямыми и парными прочтениями.
Для выравнивания использовался геном вируса Ebola по ссылке:
https://www.ncbi.nlm.nih.gov/nuccore/AF086833.2?report=genbank.
Использовались оба формата хранения генома: GNB – с аннотациями и FASTA – просто геномная последовательность.
Далее решалась задача выравнивания результатов секвенирования на геном вируса Ebola. Все выравнивания проводились на ОС Ubuntu с использованием виртуальной машины, установленной на ОС Windows 7 на компьютере с процессором Intel Core i7-2600 3.4GHz и ОЗУ 16Gb согласно инструкциям для выбранных программ и с настройками «по умолчанию».
На сайте NCBI были взяты результаты секвенирования транскиптома человека, больного лихорадкой Ebola, и геном вируса Ebola. Проводились одинаковые по содержанию, но разные по форме действия для 4-х программ выравнивания (BWA, Bowtie2, Minimap2 и Hisat2):
- установка программы,
- индексирование на геном вируса Ebola,
- выравнивание прямых прочтений,
- удаление из файла невыравненных прочтений,
- выравнивание обратных прочтений,
- удаление из файла невыравненных прочтений,
- расчёт статистики специально написанной программой сразу для двух файлов (прямых и обратных прочтений).
Общая схема работы с данными при выравнивании и его анализе
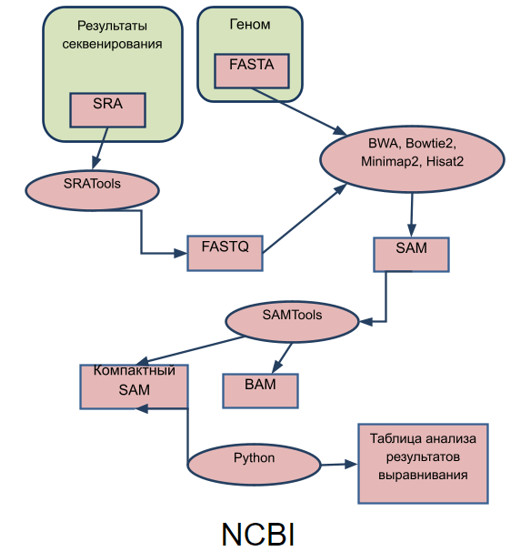
Была написана небольшая программа на языке Python, позволяющая вычислить статистику для полученных выравниваний даже без использования дополнительных библиотек для чтения форматов SAM и BAM. Также программа позволяет рассчитать статистику сразу и для прямых, и для обратных прочтений.
В работе использовались данные, полученные на секвенаторе 2-го поколения Illumina. В этом секвенаторе используется метод коротких прочтений.
Результаты
1. Данные секвенирования были удачно выравнены на геном вируса Эболы при помощи 4-х программ выравнивания.
Результаты анализа выравнивания
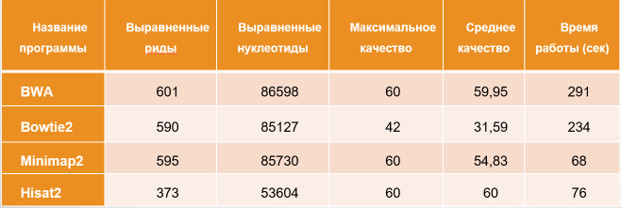
2. Программа BWA получила самое большое число выравненных ридов и с высоким качеством, но время её работы самое большое.
3. Отставание Hisat2 объясняется тем, что с настройками «по умолчанию» данная программа оставляет в выравнивании только риды с максимальным качеством.
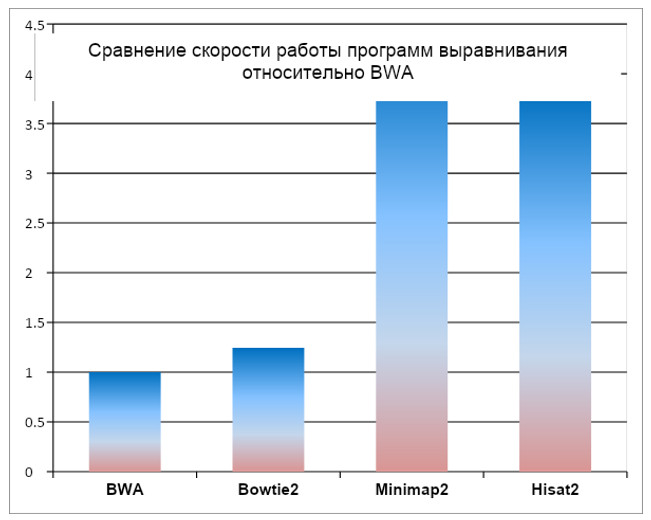
4. Программа Minimap2 значительно выделяется по скорости выравнивания и превосходит BWA в 4,3 раза. Немного отстает от Minimap2 программа Hisat2. Bowtie2 значительно медленнее, чем Minimap2.
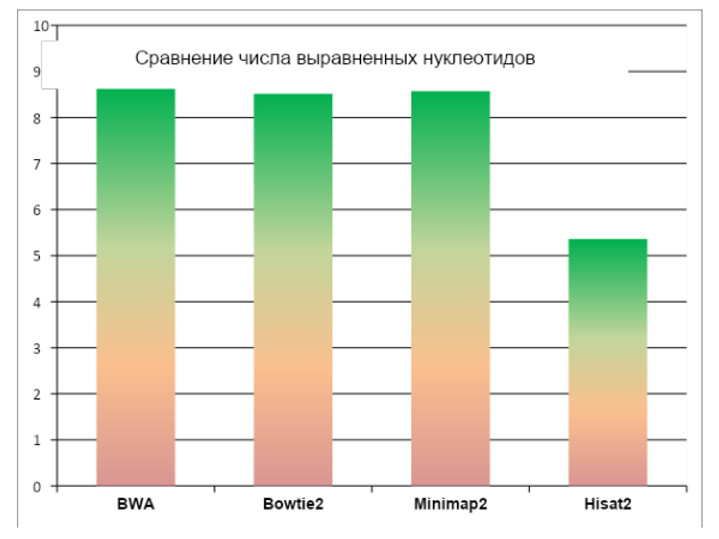
5. Программы BWA, Bowtie2 и Minimap2 выровняли примерно одинаковое количество нуклеотидов, которое с точностью до 3% соответствует таксономическому анализу, проведённому BLAST.
Выводы
1. Из исследованных программ выравнивания наибольшую производительность продемонстрировала Minimap2 при сохранении очень хорошего качества выравнивания и количества выровненных нуклеотидов.
2. Исследуемые программы затратили на выравнивание 6,7 миллионов парных прочтений на геном размером 18871 bp от 1 до 5 мин. Это становится всё более перспективным.
3. Благодаря эффективности используемых в программах выравниваний алгоритмов для решения поставленной в данной работе задачи не потребовалось больших вычислительных ресурсов.
Перспективы использования результатов работы
С использованием новых технологий скорость и точность секвенирования постоянно растёт. Метод диагностики заболеваний при помощи секвенирования с последующим выравниванием на геном становится всё более перспективным.
Награды/достижения
Международный конкурс научно-технических работ школьников «Старт в Науку» – диплом I степени.
Мнение автора
«Конкурс проектов и исследований «Старт в медицину» дал мне хороший толчок в развитии моего проекта. Я получила новый опыт, участвуя в научно-практической конференции»
- Изучение и освоение метода овогельминтоскопии для дифференциальной диагностики кишечных паразитов
- Изучение лекарственных свойств семян арбуза. Производство антигельминтного продукта для животных на их основе с применением безотходной технологии
- Изучение популяции зябликов (Fringilla coelebs) на территории Воронежского ГПБЗ имени В. М. Пескова
- Особенности нереста и выращивания скалярий
- Онтофилогенетические механизмы формирования пороков развития сердца и сосудов у человека
- Регенерация у аксолотлей амбистомы
- Бесскорлупное культивирование куриных яиц в условиях птицефермы. Современные методы и возможности их оптимизации
- Влияние неправильного питания птиц на распространение зооантропонозных заболеваний
- Влияние электромагнитного излучения сотовой связи на размножение и развитие мух Drosophila melanogaster
- Изучение прибрежной фауны жужелиц (Carabidae) как фактора биоиндикации экологического благополучия Московской области
- Изучение динамики физической активности организма в условиях светового десинхроноза
- Создание обучающего интерактивного пособия «Клещи семейств Иксодовые (Ixodidae) и Аргасовые (Argasidae) – переносчики возбудителей опасных заболеваний человека»
- Язык кошек: интересные факты и наблюдения за моим котом
- Эволюция коры головного мозга
- Влияние условий процесса СКФ-микронизации на растворимость моксифлоксацина и левофлоксацина
- Исследование чувствительности и специфичности экспрессии мРНК онкомаркера TRF1 при раке щитовидной железы
- Предотвращение изменения среза гриба
- Получение лизоцима – простейшего фермента, обладающего антибактериальной активностью. Использование в биотехнологии и медицине
- Экспрессное профилирование жирных кислот в плазме крови человека методом масс-спектрометрии МАЛДИ
- Лактазная недостаточность
- Исследование условий анализа экспрессии мРНК гена c-Myc
- Оптимизация метода ОТ/ПЦР для анализа генов MAX и MAD
- Кинезиология как гимнастика мозга
- ДНК-метилтрансфераза M.SSSI: выделение и метилирующая активность
- Синтез и анализ катионного димерного комплекса гексафторфосфат бис (циклопентадиенил-карбонилфенилтиожелеза)
- Исследование патогенеза атеросклероза на примере выборки из Московского региона
- «Кто рано встаёт, тому Бог подаёт». Миф или реальность?
- Поиск подходов к повышению эффективности амплификации ДНК фага лямбда методом ПЦР
- Изучение взаимоотношений третьих моляров нижней челюсти и нижнечелюстного канала
- Стратегия создания псевдотриглицеридных пролекарств в синтезе антиретровирусных соединений
- Определение эмоционального состояния детей дошкольного и младшего школьного возраста при помощи цвета
- Изучение свойств ферментов
- Альтернативные способы распломбировки зубных каналов
- Энергетические напитки и их влияние на организм
- Использование методов анализа уровней теломеразной активности в выявлении онкопатологий мочевого пузыря
- Изучение онкометаболитов, характерных для тканей головного мозга, с помощью методов масс-спектрометрии
- Биохимический анализ крови
- Создание пандусов нового поколения и выкатного устройства городского транспорта для людей с ограниченными возможностями на основе актуаторов
- Синтез и исследование свойств магнитных наночастиц
- Изменение скорости фотосинтеза под влиянием магнитных полей различной индуктивности
- Исследования сил
- Медиатехнологии в системе охраны здоровья граждан: создание специализированного чат-бота с алгоритмами оказания первой помощи в Telegram
- Исследование влияния наушников на слух и время реакции подростков
- Мониторинг качества родниковой воды методом биоиндикации
- Исследование теплопроводности мягких тканей человека на фантоме из пластизоля методами пассивной акустической термометрии и инфракрасной термографии
- Анализ остроты зрения учащихся
- Подбор условий и сред для оптимального набора биомассы бактерий при продукции белка
- Использование метода регистрации светорассеяния для изучения агрегации белков
- Создание прибора для определения гипермобильности мизинца, локтевого и коленного суставов
- Исследование методов цифровых отображений био-физико-химических параметров молекулярно-генетического кода
- Анализ влияния кондиционной среды мезенхимальных стволовых клеток костного мозга на течение токсического поражения печени при действии парацетамола
- Протезирование костей человека
- Экспресс-способ оценки антибиотикорезистентности бактерий
- Выравнивание результатов секвенирования на геном вируса Ebola
- Полиморфизм гена MC1R
- Система сканирования результатов обследования пользователя с целью выявления диагноза
- Создание части карапакса черепахи с помощью 3D-принтера
- Эргорука
- Исследование цитоплазматической ДНК растений рода Cenchrus для создания хлоропластных маркеров на основе вариабельных STR-локусов
- Влияние состава питательной среды на морфофизиологические показатели рапса при клональном микроразмножении
- Дифференцировка по степени поражения зубных рядов у лиц возрастной категории 30–40 лет
- Культура клеток высших растений. Урок-лекция для учащихся 9–11 классов
- Криоконсервация продуктов функциональной направленности на примере творога
- Содержание углекислого газа в помещениях школы в условиях длительного эксперимента
- Влияние противогололёдных реагентов на почву и растения
- Влияние дистанционного обучения на зрение
- Содержание пищевых добавок в йогуртах
- Сердечно-лёгочная реанимация в рамках первой помощи: готовность оказывать и желание изучать учениками старших классов
- Изучение ассортимента и оценка перспектив использования мужских контрацептивных препаратов в России
- Инновационный метод борьбы с синантропной флорой транспортных путей на примере борщевика Сосновского с помощью растений-ремедиаторов
- Изучение влияния искусственного ультрафиолетового излучения на жизнедеятельность живых организмов
- Cравнительный анализ эффективности различных видов масок и респираторов, применяемых в качестве средств индивидуальной защиты
- Репеллент для животных на основе растительного сырья
- Определение лактата и мочевины в кожном экскрете
- Влияние энергетических напитков на организм человека
- Влияние состояния микросна на эффективность запоминания лекционной информации в виртуальной среде
- Исследование связи между воздействием на вестибулярный аппарат человека и изменением частоты нистагма глаз
- Оценка психомоторного развития детей с нарушениями опорно-двигательного аппарата
- Взаимосвязь болевого синдрома и веса у детей с тяжёлыми хроническими заболеваниями
- Оценка точности методик определения возраста на основании морфологии зубов на основе развития второго моляра. Метод «Moorees, Fanning and Hunt»
- Биоэлектрическая активность сетчатки при миопии разной степени
- Возможности коррекции расстройств сна посредством тренингов с использованием биологической обратной связи
- Оптимизация психофизиологического состояния спортсменов старшего подросткового возраста
- Дистанционное обучение и здоровье человека
- Мониторинг физического развития обучающихся
- Анализ методик, используемых для адаптации и социализации детей с синдромом Дауна
- Сравнительный анализ параметров альвеолярного гребня челюстей у людей при потере зубов
- Половые различия в мотивации и особенностях употребления энергетических напитков старшеклассниками
- Выявление факторов риска развития сердечно-сосудистых заболеваний у подростков и оценка их влияния на когнитивные способности, стрессоустойчивость и функциональное состояние сердечно-сосудистой системы
- Сравнительная оценка методик определения возраста человека, основанных на оценке развития корней третьих моляров
- Вальгусная деформация первого пальца стопы
- Разработка специальной одежды для людей с ОВЗ
- Исследование эмоционального состояния учащихся при дистанционном обучении
- Разработка методического пособия по изучению латинского языка для учащихся медицинских образовательных учреждений
- Влияние дистанционного обучения на здоровье учителей, школьников и студентов
- Разработка методического обеспечения открытого урока на тему «Формирование сексуальной грамотности как необходимый элемент воспитания подростков»
- Средства смягчения высказывания в медицинской речи в художественных текстах русской литературы
- Выявление учеников 6-х классов школы № 138 г. Москвы, испытывающих проблемы во взаимоотношениях с одноклассниками
- Внимание как познавательный процесс и стратегии достижения его максимальной концентрации
- Исследование взаимосвязи перфекционизма и формирования системы ценностей учащихся 10-х классов
- Оценка эффективности правил общения с ребёнком-аутистом (по Никольской) на примере отдельного случая
- Зависимость наличия признаков заболеваний сердечно-сосудистой системы от вида трудовой деятельности
- Изменение ритмической активности нейронов при формировании моторного навыка
- Разработка системы мониторинга и профилактики артериальной гипертензии
- Влияние уровня тревожности на когнитивные функции и вегетативную нервную систему учеников 10-х классов
- Изучение влияния разных музыкальных жанров на сердечно-сосудистую систему подростков
- Влияние режима сна на развитие тревожности и успеваемость учащихся подросткового возраста
- Синтез аналога радиофармпрепаратов на основе металла галлия и хелатора тетракарбоновой кислоты (DOTA) с потенциальной визуализирующей и противоопухолевой активностью
- Влияние стимуляции определённых эмоций учащегося старших классов на результативность его учебной деятельности / The interaction between stimulated students` emotions and their academic results
- Исторический опыт: разработка показателей качества сырья манго (Mangifera indica) / Historical experience: analysis of quality indicators of raw mango (Mangifera indica)
- Конструкционные стоматологические материалы в зеркале истории / Prosthetic materials in the mirror of history
- Исторический опыт: фитохимический анализ отходов переработки плодов перца сладкого стручкового однолетнего (Capsicum annuum L.) / Historical experience: phytochemical analysis of waste products from the fetus of the bell pepper (Capsicum annuum L.)
- Исследование макрофауны беспозвоночных животных реки и пойменных водоёмов Лихоборки
- История кафедры анатомии МГМУ им. И. М. Сеченова / The history of the department of anatomy MGMU im. I. M. Sechenova
- История медицинских открытий в годы Первой мировой войны / History of medicine and medical advances made during World War one
- Отсутствие исторической памяти приводит к ошибкам: как возникла эпидемия кори в 21 веке? Исследование на примере коллектива школы № 1535 / Historical amnesia leads to tough mistakes: how measles epidemic is possible in the 21st century?
- Вирус бешенства в истории человечества. История создания первой вакцины / Rabies virus in the history of mankind. The first vaccine
- Из истории лекарств. Анестетики / From the history of medications. Anesthetics
- Биоиндикация водоёмов Измайловского парка по комплексу Протистофауны
- Качественный анализ аминокислот методом хроматографии
- Синтез комплекса на основе золота и тетрафенилпорфирина (TPP) с потенциальной фотодинамической антибактериальной активностью
- Выявление эколого-биологических особенностей адаптации вольфии бескорневой к действию абиотических и антропогенных факторов в различных условиях культивирования in vitro
- Зинаида Виссарионовна Ермольева: вклад в развитие медицины и медицинской помощи в период Великой Отечественной войны / Zinaida Vissarionovna Yermolyeva: contribution to the development of medicine and medical care during the Great Patriotic War
- Синтез производного глутатиона
- Выбор растворителей для реакции получения эпихлоргидрина на основе исследования их свойств
- Фармакогностическое изучение сырья вишни обыкновенной Cerasus vulgaris Mill.
- Изучение сформированности принципов здорового питания у школьников подростков
- Защитные свойства зубных паст
- Профилактика снижения зрения у подростков в современном digital мире
- Причины и профилактика кариеса у учащихся начальных классов
- Сравнительная оценка выявления зубного налёта в домашних условиях
- Исследование эффективности и безопасности различных форм антиперспирантов, изготовленных в домашних условиях и на производстве
- Поиск новых генов и регуляторных механизмов, отвечающих за формирование и поддержание статуса предшественников половых клеток Drosophila melanogaster
- Оценка эффективности определения генетических полиморфизмов, ассоциированных с высокими спортивными достижениями
- Изучение метода выделения ДНК из биологического материала в условиях школьной лаборатории
- Создание растений-биосенсоров на тяжёлые металлы
- Исследование влияние мутации гена white на продолжительность жизни Drosophila melanogaster
- Влияние эндогенных активных форм кислорода на репродуктивную систему дрозофилы
- Применение экспериментальных моделей лабораторных животных в медицине. Исследование яичников самок линии иммунодефицитных мышей Balb/c nude
- Создание леденцов с жидким экстрактом листьев элеутерококка колючего (Eleutherococccus senticosus Мaxim)
- Изучение концентрации внеклеточной ДНК и нуклеазной активности у женщин с нормально протекающей беременностью, преэклампсией и гестационной артериальной гипертензией
- Сравнительный анализ антибактериальной активности ряда пряных растений (аира, куркумы и имбиря) и оценка перспектив их использования в медицине
- Фармакогностическое изучение листьев котовника кошачьего с целью расширения сырьевой базы
- Тетродотоксин
- Влияние компьютерных игр на процессы обучения учеников начальной школы с разным уровнем развития
- Справочник для слышащих по способам общения с глухими и слабослышащими
- Наука прошлого. От анатомии до робототехники. Механический барабанщик Леонардо да Винчи
- «Звёздная сыпь» в зеркале мировой литературы
- Больничное радио и его роль в психологическом сопровождении онкопациентов
- Медицинская служба во время Великой Отечественной войны
- Факторы, влияющие на психическое здоровье человека на примере анализа биографии и творчества русского художника М.А. Врубеля (1856-1910) и норвежского живописца Э. Мунка (1863-1944)
- «BrainFitness» - мнемотехники и скорочтение
- Основные причины депрессии у подростков
- Герои картин Третьяковской галереи как атлас заболеваний своего времени
- Разработка экспозиции и экскурсии медицинской службы Красной Армии в период Великой Отечественной войны 1941-1945 в школьном музее «Северный флот»
- Перфекционизм как патологический феномен
- Химия – просто! История создания одной компьютерной игры
- Стресс. Его влияние на организм. Способы снятия стресса
- Разработка методов оценки психологического состояния школьника в период повышенных психоэмоциональных нагрузок при подготовке к сдаче основного государственного экзамена
- Типы когнитивной активности при понимании текста (на материале научно-популярного текста)
- Исследование взаимного действия ингибиторов на каскад коагуляции
- Оптимизация условий анализа экспрессии сплайсинговых вариантов мРНК гена сурвивина методом ОТ/ПЦР
- Изучение влияния компонентов клеточной стенки дрожжей Saccharomyces cerevisiae на фибриллизацию инсулина
- Ферментные цитотоксические препараты для терапии опухолевых заболеваний
- Анализ уровня теломеразной активности в лизатах опухолевых клеточных линий SKOV3 и K562
- Исследование уровня экспрессии протоонкогена с-Myc в клинических образцах новообразований щитовидной железы
- Оптимизация условий анализа мРНК генов MAX и MAD с целью определения активности этих генов методом ОТ/ПЦР
- Шапероны человека в клетках кишечной палочки
- Исследование ингибирующего влияния ряда фосфоротиоатных олигонуклеотидов на активность теломеразы в лизатах опухолевых клеток MCF-7 in vitro
- Анализ уровней теломеразной активности методом TRAP и методом TRAP/PCR в реальном времени (RT-TRAP-2PCR)
- Разработка технологии получения полиэтиленового вкладыша тазобедренного сустава с биоактивным пористым слоем
- Магнитное поле как фактор управления морфологией растений
- Изучение аудиометрии как одного из наиболее эффективных способов определения тугоухости
- Прибор для отслеживания уровня радиации и концентрации опасных для человека газов
- Создание модели экзоскелета руки
- Криоконсервация семенной жидкости в растворе метанол-вода
- «Сириус» – тифлоприбор с датчиками и приложением для слепых и слабовидящих людей
- Создание обучающей модели для слабовидящих и незрячих, помогающей в изучении шрифта Брайля и алфавита
- Смерть Базарова: досадная случайность или трагическая закономерность
- Оптимизация метода FISH на бактериях
- Разработка и получение аналога геля-венотоника в лабораторных условиях
- Изучение гена FRIGIDA-LIKE в видах Brassica
- Косметические спиртосодержащие лосьоны для ухода за кожей лица
- Коррекция функций кардиореспираторной системы средствами танцевальной терапии у детей дошкольного возраста с кохлеарной имплантацией
- Сравнение коррозионной стойкости аморфного и кристаллического сплава на основе циркония
- Исследование действия препарата «Мексидол» на показатели поведения лабораторных крыс
- Создание косметического средства на основе алоэ вера и аллантоина
- Особенности вегетативной реактивности студентов при проведении тепловой пробы
- Определение коэффициента водопоглощения лекарственного растительного сырья корневищ лапчатки прямостоячей и листьев эвкалипта
- Качественное и количественное определение салициловой кислоты в медицинских препаратах и изучение её свойств
- Сравнительный анализ подлинности и количественного содержания лютеина в биологически активных добавках (БАД) к пище различных производителей методом тонкослойной хроматографии и спектрофотометрии
- Синтез органических роданидов и определение их противогрибковой и противобактериальной активности
- Метод твёрдых дисперсий – перспективный способ повышения растворимости малорастворимых в воде лекарственных веществ на примере метронидазола
- Создание спрея от клещей и насекомых на основе натуральных растений репеллентов: пижмы (Tanacetum) и полыни (Artemisia)
- Перспективы использования ферментированного раствора чайного гриба (Medusomyces gisevii) в фармацевтической промышленности
- Непереносимость нестероидных противовоспалительных препаратов. Аллергия на анальгин и возможный альтернативный препарат
- Анализ номенклатуры и ассортимента зубных паст, представленных в РФ. Разработка состава зубной пасты с экстрактом кожуры апельсина, обладающей антимикробной и противовоспалительной активностью для профилактики воспалительных заболеваний пародонта
- Влияние эпигенетических модуляторов С-646 и А-485 на секрецию внешней протеазы активатора протеина С и Х фактора
- Химические свойства глюкозы и патологические состояния, связанные с нарушением её метаболизма в организме
- Выбор и оптимизация условий ОТ/ПЦР для высокоэффективного анализа уровня экспрессии мРНК гена C-MYC
- Лекарственная плёнка пролонгированного действия с витамином B6 и глицином: способы изготовления и применения
- Разработка состава и технологии получения шипучей таблетки с экстрактом черники
- Изучение свойств и влияния на здоровье человека воды из святого источника села Дубовое Липецкой области в сравнении с водой из других источников (родника, колонки, реки) данного села
- Колориметрическое определение железа общего в водоёмах деревни Анциферово Орехово-Зуевского района Московской области
- Фитохимический анализ изучения возможностей комплексной переработки сырья рябины обыкновенной (Sorbus aucuparia fructus)
- Получение эфирных масел из растительного сырья и сравнительный анализ эфирных масел различного производства
- Синтез нового экологически чистого сорбента на основе яблочного пектина
- Исследование поглотительной способности энтеросорбентов по отношению к катионам тяжёлых металлов
- Разделение антибиотиков ванкомицина гидрохлорида и эремомицина сульфата с помощью метода гидрофильной хроматографии
- Количественное определение содержания цианокобаламина в лекарственных препаратах и БАДах
- Использование метода поляриметрии для оценки эквивалентности взаимозамены лекарственных препаратов на основе глюкозы
- Исследование физико-химических свойств шампуней
- Лимонен – только ли польза?
- Изучение оптической активности веществ
- Разработка состава и технологии получения лекарственного препарата в форме спрея на основе мумиё для лечения воспалительных заболеваний полости рта и горла
- Разработка состава и технология приготовления облепиховой мази для лечения стоматита в лаборатории и домашних условиях
- Изготовление карамели с календулой лекарственной для профилактики и лечения заболеваний полости рта
- Создание нового комбинированного противовоспалительного препарата из лекарственного сырья растений, смолы которых входили в рецептуру лекарства, описанного в текстах Ветхого Завета
- Перспективы создания экстракционного препарата из плодового тела шампиньона двуспорового (Agaricus bisporus)
- Сахарный диабет: заболевание XXI века или образ жизни?
- Преимущества выращивания мяты перечной методом водной культуры. Применение свойств масла мяты в медицине
- Оптимизация процессов синтеза комплекса гадопентетовой кислоты, используемого в качестве контрастного диагностического препарата для магнитно-резонансной томографии
- Расторопша и её значение в сфере медицины
- Возможности введения базилика камфорного в список фармакопейных лекарственных растений
- Изучение и разработка показателей качества плодов Myristica fragrans Houtt
- Фитохимический анализ и изучение перспектив использования в медицине листьев пеларгонии
- Лекарственные растения как источники получения масел. Перспективы и возможности на примере кунжута
- Выведение цыплят в скорлупе и в бесскорлуповой оболочке в искусственно созданных условиях среды
- Оценка перспектив использования растения Толокнянка обыкновенная (Arctostaphylos uva-ursi) для лечения симптомов постакне
- Исследование влияния температурного фактора на количество числа позвонков в осевом скелете популяции щуки обыкновенной
- Изучение характеристик подлинности травы амаранта запрокинутого
- Животные-целители
- Репаративная регенерация (эпиморфоз) на примере аннамского палочника (Medauroidea extradentata)
- Расширение базы микропрепаратов по зоологии в школьном кабинете биологии
- Фармакогностический анализ и создание лекарственного средства на основе сырья кипрея узколистного (Chamaenerion angustifolium L.)
- Улитка-антистресс. Неврологические аспекты улиткотерапии
- Анализ перспектив изучения листьев и плодов винограда культурного для составления нормативной документации на новые виды ЛРС
- Фармакогностическое изучение сырья розмарина лекарственного (Rosmarinus officinalis L.)
- Исследование состава и полезных свойств листьев аронии черноплодной (Aronia melanocarpa (Michx.) Elliott). Перспективы их использования в производстве биологически активных добавок, разработка фиточая
- Лабораторные этапы изготовления непрямых ортопедических конструкций с использованием CAD/CAM
- Влияние акне на жизнь подростков
- Сельдь как источник заражения анизакидозом
- Разработка методов лабораторной диагностики Blastocystis Species и изучение роли домашних животных в инфицировании человека бластоцистами
- Биополимер на основе крахмала
- Регистрация и анализ отличий паттернов поведения в норме и при остром абстинентном синдроме у лабораторных крыс стока Wistar
- Влияние глутаминовой кислоты на интенсивность выработки паутинного секрета у пауков (на примере Brachypelma albopilosum)
- Перспективы применения продуктов вторичной переработки на примере кофейных оболочек при производстве хлебобулочных изделий с целью увеличения качества продукции
- Изучение филогенетических особенностей черепа человека с применением объёмной фотографии
- Исследование микробиологической обсеменённости предметов, часто используемых человеком
- Получение экстракта слизи улиток и его использование для внеклеточного матрикса
- Влияние йода на развитие проростков овса посевного (Avena sativa L.)
- Изучение биологических плёнок бактерий на абиотических поверхностях
- Экосистемы лесов окрестностей биогеоценологической станции «Малинки»
- Получение активированного углеродного материала путём термической переработки растительного сырья на примере рисовой шелухи
- Исследование цитопатического действия вируса паротита на культуру клеток меланомы человека
- Биомониторинг северной акватории полуострова Большой Утриш
- Разработка аналогового действующего вещества препаратов для лечения и профилактики железодефицитной анемии
- Морфологический анализ структуры лёгкого в экспериментальной модели эндотоксического воздействия липополисахарида (инфекционно-токсический шок) на дыхательную систему крысы
- Влияние абиогенных факторов на развитие рода Penicillium
- Анализ лекарственных форм и биологически активных веществ на содержание иодида калия, ионов кальция и магния
- Получение моноклональных антител против антигенов микобактерий из Micobacterium tuberculosis complex
- Качественный состав зубных паст разных производителей, их сравнение и влияние лучшей на организм человека
- Прибрежная фауна жужелиц (Carabidae) водоёмов в с. Павловская Слобода и деревне Козлово Московской области
- Мониторинг качества воздуха в районе Гольяново по снеговому покрову на примере территории школы № 1516
- Использование ореха грецкого в стоматологии
- Эффективность применения защитного раневого покрытия у пациентов в послеоперационном периоде
- Исследование проявления асимметрии листа берёзы повислой в онтогенезе
- Распространение, численность и роль панцирных клещей
- Денисовский человек и его роль в эволюции современного человека
- Исследование бактериальной обсеменённости поручней в метро, изготовленных из различных материалов. Исследование средств для дезинфекции поручней и деконтаминации рук
- Сравнительная характеристика морфологических и экологических особенностей некоторых представителей орнитофауны Подмосковья и Крыма
- Сравнительный анализ содержания натрия, калия и железа в проростках различных культур
- Разработка нового регенерирующего средства на основе йода и серебра
- Изучение влияния аскорбатоксидазы на содержание витамина С во фруктово-овощных смесях
- Влияние эфирных масел на микроорганизмы
- Близорукость у школьников. Её причины и профилактика. Влияние современных средств коммуникации на формирование органа зрения у школьников
- Влияние уборки листового опада в городе на почвенную мезофауну
- Исследование влияния эфирных масел на рост микроорганизмов
- Влияние ампициллина разной концентрации на живые организмы
- Инфузории активного ила – индикаторы степени очистки сточных вод
- Влияние современных гаджетов на зрение школьников ГБОУ Школа № 1788 г. Москвы
- Распространённость заболеваний полости рта у жителей Республики Калмыкия
- Анализ факторов риска сахарного диабета второго типа у населения разных возрастных групп
- Получение бактерий, синтезирующих белок лёгкой цепи нейрофиламентов человека (NFL)
- Изучение возможностей получения адсорбирующих средств из древесно-растительных отходов
- Влияние акустических гаджетов на слух подростка: гигиена использования акустических гаджетов
- Моноклональные антитела как современный подход к изучению вируса гепатита C
- Влияние мочевины, содержащейся в жевательных резинках, на поддержание кислотно-щелочного баланса ротовой полости и определение её методом уреазной пробы
- Качественный анализ шоколада
- Получение сорбента на основе терморасширенного графита, модифицированного ферритами
- Исследование содержания тяжёлых металлов в грибах Солнечногорского района Московской области
- Получение химически активированного углеродного материала на основе рисовой шелухи
- Заболевание позвоночника у школьников. Причины и профилактика
- Оценка когнитивных функций и эмоционального состояния школьников с пищевой зависимостью
- Разработка биоцидного агента пролонгированного действия на основе наночастиц серебра для костного цемента
- Влияние противогололёдного реагента на окружающую среду
- Разнообразие микромицетов в декоративных насаждениях на площади Тверской заставы г. Москвы на примере яблоневых деревьев
- Влияние антибиотиков на организм при постоянном их использовании
- Книга рецептов для занятых людей
- Инновационный метод защиты городского населения от шума урбанизированных территорий
- Оценка эффективности экстракта гарцинии камбоджийской для снижения массы тела
- Качественное определение глюкозамина гидрохлорида в средах растительного и животного происхождения. Профилактика заболеваний суставов
- «Спортбокс настроения» – пропаганда регулярных физических нагрузок и ЗОЖ среди подростков
- Монилиоз. Это серьёзно?
- Вирус папилломы человека и вакцинопрофилактика
- Синдром диабетической стопы у больных сахарным диабетом: эпидемиология, мониторинг, профилактика
- Создание ассистивного устройства для слепых и слабовидящих людей
- Гендерные различия в моделях образа жизни современных старшеклассников
- Влияние характера питания на развитие кариеса у детей
- Невидимая опасность среди нас
- Вирус папилломы человека: знания учениц школы № 138 о возможностях вакцинопрофилактики
- Влияние синтетических моющих средств на здоровье человека
- Исследование свойств зубных паст
- Образ кофе в общественном сознании
- Характеристика точности методов определения активности амилазы слюны в зависимости от рациона питания испытуемых
- Воздействие аллергена луговых трав на поведенческие особенности мышей
- Гигиена зрения. Диагностика и профилактика
- Исследование феномена специализации нейронов
- Взаимосвязи между гипоксической устойчивостью, астеническими состояниями у детей и психологическими характеристиками (Опросник детской тревожности CMAS)
- Оптимизация бытового использования пластических масс
- Позитивное и негативное влияние мобильных телефонов на жизнь и здоровье подростков
- Компьютерная морфометрия в изучении межполушарной асимметрии мозга человека
- Сравнение конфокальной лазерной флуоресцентной и световой микроскопии
- Влияние нефтепродуктов и тяжёлых металлов на состав почвы вдоль магистралей Москвы
- Взаимодействие между гипоксической устойчивостью и реактивностью сосудов микроциркуляторного русла
- Внеклеточная ДНК и количество митохондриальных повторов при преэклампсии и гестационной артериальной гипертензии
- Исследование влияния рабочего места на появление профессиональных заболеваний у работников локомотивных бригад
- Повреждение сперматогенеза и перестройка эндокринного аппарата семенника мышей-гибридов CBA×C57Bl6 при действии эндотоксина клинического штамма Salmonella enteritidis
- Сравнительный анализ содержания акриламида в чёрном кофе в зависимости от степени обжарки зёрен
- Оценка фактического питания детей школьного возраста в соответствии с их соматотипами
- Экология Шестого Океана «ЭШО v.2.3»
- Изделия из нагреваемого табака. Оценка рисков потребления в сравнении с обычными сигаретами
- Оценка безопасности и качества пищевых продуктов по содержанию влаги и сухих веществ
- Разработка методики преодоления синдрома хронического стресса, обеспечивающей снижение уровня адреналина физическими нагрузками
- Влияние различных факторов на силу мышц кисти руки
- Понятие морфологии человека: гистологический метод исследования тканей человека
- Влияние рациона питания на состав мочи. Исследование кислотности и осадка мочи на примере вегетарианской и мясной диет
- Изучение влияния питания с низкой гликемической нагрузкой и аэробных физических нагрузок на объективные показатели состояния организма как форма диагностики старения
- Исследование соответствия наилучшего стиля плавания пловцов их антропометрическим данным
- Изучение факторов, влияющих на эффективность выявления антител к возбудителям Лайм-боррелиоза на иммуночипе
- Изучение особенностей вегетативного обеспечения деятельности организма учеников 9-х классов посредством проведения ортоклиностатической пробы
- Применение инструментальных методов регистрации актуализации субъективного опыта с целью выявления скрываемой информации
- Молекулярная диагностика рака предстательной железы на основе анализа рецепторов фактора роста сосудов
- Взаимосвязь между гипоксической устойчивостью организма человека и показателями вариабельности сердечного ритма (временными и спектральными)
- Изучение видового состава эпифитных лишайников городского округа Серпухов в целях выявления эффективных биоиндикаторов и разработки методики оценки состояния окружающей среды с их помощью
- Влияние тренингов с использованием биологической обратной связи на степень выраженности симптомов вегетативного дисбаланса
- Индивидуальная частота максимального альфа-пика и способность к монотонной деятельности
- Уровень внушаемости учащихся средней школы как предиктор успешности обучения
- Особенности организации поведения в зависимости от ожидаемых результатов на примере решения математических задач
- Влияние уровня тревожности на параметры сенсомоторной реакции
- Анализ общей выживаемости как метод изучения эффективности лечения больных в онкологии
- Изучение влияния факторов внешней среды на рост человека
- Каталог проектных и исследовательских работ участников, победителей и призеров открытых городских научно-практических конференций 2019 года
- Разработка импланта для краниопластики черепа
- Оценка качества ромашки лекарственной, реализуемой в аптечных сетях города Москвы
- Составление определителя млекопитающих
- Исследование бактерицидных свойств антисептических средств для лошадей
- Использование низкотемпературной плазмы для уничтожения антибиотикорезистентных штаммов микроорганизмов
- Режим дня как основа здорового образа жизни
- Травяные палы как катастрофический фактор снижения биоразнообразия, изменения свойств почвы и микробиологической активности
- Исследование микробиологической обсеменённости пластиковых карт, бумажных денежных купюр и монет
- Изменение клеточного состава грудного молока в зависимости от периода лактации и срока хранения
- Использование сидератов для обогащения почвы азотом и их влияние на содержание нитратов в овощах
- Выявление генетического материала Mycobacterium leprae с помощью различных тест-систем методом ПЦР в реальном времени
- Разработка батончика для профилактики атеросклероза с помощью растительных продуктов, имеющих лечебные свойства
- Антоцианы – экологичные индикаторы и полезные пищевые красители
- История развития нетрадиционной медицины калмыцкого народа
- Всестороннее изучение и сравнительный анализ современных шовных материалов с возможностью совершенствования нити хирургической
- Аугментация костной ткани в стоматологии с применением синтетического биоматериала
- Изучение действия различных групп антибиотиков на микроорганизмы. Антибиотикорезистентность
- Влияние диеты с повышенным содержанием аскорбиновой кислоты на структурную функцию NADPH-оксидазы группы DUOX в линиях мух Drosophila melanogaster
- Определение активности моноклонального антитела 6В11 к рекомбинантному белку вируса гепатита С методом ИФА
- Мир микроорганизмов на наших гаджетах
- Исследование антимикробных свойств зубных паст и ополаскивателей для полости рта
- Прокрастинация среди подростков поколения «М»: причины возникновения, связь с тревожностью и методы её преодоления
- Обоснование показателей качества и изготовление капсул с сухим экстрактом слоевищ цетрарии исландской (Cetraria islandica (L.) Ach.)
- Ахатина в вашем доме
- Исследование взаимодействия ДНК-аптамерных ингибиторов с тромбином
- Рекомендации для подростков по питанию вне школы
- Изучение экологической обстановки в метрополитене
- Оценка качества воды Княжегубской ГЭС Кандалакшского района Мурманской области по составу зообентосных организмов
- Анализ сточных вод фармацевтического производства методом поляриметрии
- Помощь в определении нехватки и избытка витаминов в организме человека
- Исследование качественного и количественного состава святой воды
- Создание прототипа «умного» аквариума
- Биомониторинг состояния биоценозов Измайловского парка
- Исследование качества бутилированной воды различных производителей
- Действие фитонцидов комнатных растений на простые организмы на примере инфузории-туфельки
- Разработка метода выделения белка из растительных продуктов
- Формирование наносистем на основе наночастиц серебра, стабилизированных крахмалом, и их применение
- Разнообразие и изменчивость питания обыкновенной пустельги (Falco tinnunculus)
- Зависимость эффективности трансформации бактерий относительно размера плазмиды
- Корь: знания учеников и учителей школы № 138
- Тифлоприбор «Сириус» для обучения ориентированию в пространстве слепых и слабовидящих людей
- Разработка лака на основе наночастиц серебра для защиты компьютерной клавиатуры от микроорганизмов
- Сравнение влияния средств для чистки полости рта на кальцинированный объект
- Анализ препаратов на основе животного сырья
- Применение бактериофагов для профилактики послеоперационных раневых инфекций
- Подбор оптимальных условий проведения ОТ/ПЦР для генов MAX и MAD с целью определения уровня экспрессии их мРНК
- Искусственные экосистемы. Роль в передаче инфекционных заболеваний на примере болезни легионеров
- Модель нимфы самки Dermacentor sp., особенности ротового аппарата
- Особенности регенерации белой планарии в различных условиях среды
- Изменение элементного состава минерализата под воздействием буферных растворов
- Исследование эффективности фотодинамической терапии с использованием природных фотосенсибилизаторов и коллоидного раствора наночастиц золота
- Использование магнитного поля для управления морфологией растений
- Отражение проблемы морфинизма в художественной литературе
- Шунгит - природный фильтр
- Изучение возможности корреляции функциональных особенностей сердечно-сосудистой системы и эмоциональным состоянием учеников параллели 11-х классов ГБОУ Школы № 2005 посредством проведения Гарвардского степ-теста
- Заболевания костей
- Оценка эффективности антидепрессивной терапии у больных с аффективной патологией на основании фармакогенетического исследования
- Что такое вейпинг и его влияние на здоровье подростка
- Выбор и оптимизация условий ОТ/ПЦР для высокоэффективного анализа уровня экспрессии мРНК гена c-MYC
- Полиморфизмы гена МАОА как предикторы личностных диспозиций
- Применение компьютерного зрения совместно с ультразвуковым методом ориентации в пространстве для слепых и слабовидящих людей
- Оценка качества воды озера Ковдозеро в окрестностях посёлка Зеленоборский Кандалакшского района Мурманской области методом флуктуирующей асимметрии окуня обыкновенного (Perca fluviatilis)
- Современное эндопротезирование суставов
- Оптимизация процесса культивирования эукариотических клеток-продуцентов линии СНО при производстве моноклонального антитела PRK
- Фитохимическое изучение и анализ перспектив использования сырья мушмулы (Muspilus), интродуцированной в Российской Федерации
- Укрепление ногтей акриловой пудрой
- Изучение возможности получения экстракционного препарата из плодового тела белого гриба (лат. Boletus edulis Bull)
- Изучение окраски уличных кошек
- Сонификация фотоэффекта как средство отображения информации в тифлопедагогике
- Изучение состава горького шоколада и сравнение с нормативным документом − ГОСТом
- История развития компьютерной томографии как диагностического метода
- Анализ ассортимента лекарственных средств и оценка перспектив создания повязки с экстрактом крапивы двудомной (Urtica dioica L.) ранозаживляющего действия
- Проектирование велосипеда для инвалидов без верхних конечностей. Классификация и изучение оборудования для инвалидов
- Фармакогностическое изучение сырья хризантемы (Сhrysanthemum L.)
- Сравнительный анализ интродуцированных на Украину видов скальных ящериц рода Darevskia с использованием микросателлитного маркирования
- Изучение возможности применения интерактивных измерительных устройств в краниологических исследованиях
- Современные методы диагностики болезни Альцгеймера на ранних стадиях
- Анализ свойств некоторых молочных продуктов в зависимости от длительности их сроков годности
- Виды доброкачественных мезенхимальных опухолей тканей человека. Патологоанатомическая диагностика их на примере доброкачественной опухоли матки
- Влияние напитков разного pH-показателя на зубную эмаль
- Исследования лазерной кристаллизации, химического и механического упрочнения коллагена и хитозана, улучшение залечивания повреждённых биотканей
- Первая помощь в школе: желание изучать и оказывать
- Влияние информационных перегрузок на качество жизни современного человека
- Изучение возможности получения косметического средства на основе экстракта из слоевища Пармелии блуждающей (Parmelia vagans Nyl.) для профилактики акне
- Аутоплазмозамещение у больных диффузным токсическим зобом
- Порог вкусовой чувствительности к глюкозе как способ диагностики предрасположенности к нарушениям углеводного обмена
- Загрязнение и фитотоксичность снежного покрова города Москвы на примере района Ленинские горы
- Сравнительный анализ качественных реакций, используемых для идентификации и обнаружения примесей по различным фармакопеям, на примере метенамина
- Изучение распространённости поллиноза среди старшеклассников
- Трёхмерная модель лактозного оперона Escherichia coli
- Исследование содержания ионов железа в яблоках
- Определение общего содержания антиоксидантов в соках методом амперометрического детектирования
- Изменение анатомического строения хвои Рinus sylvestris (L.) в условиях антропогенной нагрузки на территории СВАО города Москвы
- Гигиена воздуха в учебных кабинетах школы
- Знакомый и незнакомый йодат калия
- Теоретические и экспериментальные аспекты разработки различных лекарственных форм на основе полоксамеров
- Анализ показателей качества и оценка перспектив комплексной переработки плодов и листьев айвы
- Подростки и плоскостопие
- Daucus carota или морковь обыкновенная как антиоксидант и биологически активный продукт
- Акцентуации характера подростков в современном обществе
- Анализ уровня экспрессии генов VEGF и его рецепторов KDR/FLT-1 методом ОТ/ПЦР в образцах клеточной РНК из биоптатов пациентов с новообразованиями предстательной железы
- Йододефицитные заболевания в Воронежской области: эпидемиология, мониторинг, профилактика
- Особенности развития навыков письма у учеников первого класса с разным уровнем психомоторного развития
- Разработка метода сбора и концентрирования биомассы
- Исторический опыт и перспективы использования ионов цинка в медицине
- Информационно-коммуникационные технологии на уроке химии
- Плазмолиз как метод лиофилизации при холодном анабиозе
- Изучение влияния алкоголя и низкоинтенсивного излучения лазера на активность тромбоцитарного звена гемостаза
- Влияние различных способов обработки на прорастание твердокаменных семян Галеги восточной с целью увеличения их всхожести для решения экологических аспектов продовольственной безопасности
- Влияние регуляторов роста растений на пшеницу в условиях различных типов засолений
- Дубильные вещества, их классификация. Особенности стандартизации сырья, содержащего дубильные вещества. Особенности использования в современной медицине
- Исследования лазерной кристаллизации, химического и механического упрочнения коллагена и хитозана
- Анализ атмосферного воздуха в некоторых городах России по чистоте снежного покрова
- Исследование экологического состояния водоёмов Новоуральского городского округа методом оценки стабильности развития морфологических признаков плотвы обыкновенной (Rutilus rutilus) и окуня речного (Perca fluviatili)
- Зелёная энергетика
- Диспластические изменения тазобедренных суставов у людей и крупных пород собак
- Исследование видового состава флуоресцентной микрофлоры язвенных повреждений кожных покровов дальневосточной горбуши
- Использование вирусных векторов для продукции белка в растении: от модельного GFP к целевому иммуноглобулину G
- Картирование окрестностей деревни Саргая Бурзянского района республики Башкортостан
- Выращивание листового салата методом гидропоники в зимнее время
- Исследование уровня шума авиационного и железнодорожного транспорта в поселении Рязановское
- Влияние занятий воркаутом (STREET WORKOUT) на здоровье подростков и молодёжи
- Минеральные сорбенты – природные фильтры для очистки экосистем и организма
- Перспектива использования мха сфагнума для производства биологически активных добавок (БАДов) на территории РФ
- Синтез и физико-химические свойства графитовой фольги модифицированной фосфор- и борсодержащими антиокислительными добавками
- Есть для того, чтобы жить
- Биологическое производство растительных волокон с заданными физико-химическими свойствами
- Использование растительных пигментов как альтернативы синтетических кислотно-основных индикаторов
- Определение токсичности воды в водоёмах Измайловского лесопарка методом биотестирования с использованием культуры ракообразных Ceriodaphnia affinis
- Комплексные и композиционные полисахаридные криоструктураты биомедицинского назначения
- Создание модели технологии получения гигиенического средства для очистки кожи рук
- Методический набор «Сириус» для слепых и слабовидящих детей с ОВЗ по зрению
- Экспресс-анализ проб опавшей листвы
- Исследование возможностей микробного топливного элемента на основе колонки Виноградского
- Интеллектуальная одежда: умная стелька «Пора домой»
- Разработка метода нано- и микромодификации поверхности титановых сплавов для использования в качестве имплантов
- Учебная мотивация как основной фактор успешного обучения в средней и старшей школе
- Выделение азотфиксирующих бактерий, продуцентов полигидроксиалканоатов для производства биодеградируемых пластмасс
- Изучение методов лучевой диагностики при травме костей лицевого отдела черепа
- Перспективы использования растительных антигельминтных средств
- Исследование обсеменённости бритвенных систем и подбор средств для их дезинфекции
- Медицинские аспекты использования триклозана в средствах гигиены
- Влияние витамина С (аскорбиновой кислоты) на заболеваемость ОРВИ в период сезонных респираторных заболеваний
- Антибактериальные свойства «Воды венгерской королевы Эржебет» в лечении вульгарного акне у подростков
- Хлоргидрат алюминия в антиперспирантах
- Плесень: вред или польза?!
- Культивирование одноклеточной солоноводной водоросли вида Dunaliella salina в качестве источника бета-каротина
- Калькулятор здоровья
- Изучение влияния различных антибактериальных средств на гигиену рук человека методом исследования смывов
- Исследование бактерицидной активности эфирных масел
- Введение в культуру in vitro и каллусогенез растений-продуцентов вторичных метаболитов
- Значение бактериофагов в профилактике инфекций: прошлое, настоящее, будущее
- Влияние факторов внешней среды на развитие плесневых грибов и дрожжей
- Влияние плесневых грибов на микрофлору человека
- Что такое аллергия и как ей противостоять?
- Самоорганизованность
- Проект У или Мир глазами ребёнка
- Эпигенетические нарушения сперматозоида
- Изучение микробного состава современных кисломолочных продуктов с заявленными пробиотическими свойствами и традиционных кисломолочных продуктов. их место в повседневном питании
- Проблемы организации времени учащихся как фактор их социальной успешности
- Изменчивость содержания норадреналина у активных и пассивных крыс в условиях конфликтных ситуаций
- Папиллярные узоры человека
- История искусственного сердца
- Исторический опыт изучения ассортимента, перспектив использования и стандартизации лекарственных средств на основе нафталанской нефти
- Использование приёмов мнемотехники с целью улучшения памяти человека (на примере разных возрастных групп)
- Донорство крови и её компонентов: мифы и реальность
- В фокусе Гендера – успешность
- Сравнительно-морфологический анализ ротовых аппаратов клещей семейства Ixodida
- Обыкновенный воробей
- Эпигенетика близнецов
- Нарушение гемостаза в онкогематологии
- Влияние витамина D3 на иммунную систему человека
- Исследование предрасположенности развития сахарного диабета от порога вкусовой чувствительности к глюкозе и шкалы прогнозирования сахарного диабета второго типа
- Влияние суточных биоритмов на работоспособность человека
- Некоторые аспекты, влияющие на продолжительность и успешность грудного вскармливания
- Создание измерителя пульса на основе Ардуино и изучение принципа обратной связи
- Создание образовательных приложений по биологии в формате дополненной реальности
- Молекулярно-генетическое исследование ассоциаций полиморфизма генов нейромедиаторов с интеллектуальными способностями школьников
- Мини-куры. Создание автономной мини-фермы
- Исследование «quorum sensing» системы психрофильных люминесцирующих бактерий aliivibrio logei
- Влияние режима сна на развитие тревожности у подростков
- Зоопротезирование с использованием технологии 3D-прототипирования
- Оценка конкурентных отношений между видами беломорских мидий М.еdulis и M.trossulus
- Развитие эмбрионов птиц на примере попугаев
- Составление электронного определителя растений семейства Норичниковые Башкирского государственного природного биосферного заповедника
- Неизвестная паутина хорошо знакомых пауков
- Исследование содержания биологически активных веществ в листьях и корнях «Малины обыкновенной»
- Кизил - упущение российской фармакопеи
- Влияние типа восприятия ученика на образовательный процесс
- Нанопрепарат для терапии и диагностики в онкологии
- Стандартизация биологически активных добавок на основе экстракта Босвеллии (boswellia serrata) методом ВЭЖХ-УФ
- Исследование листьев крыжовника отклонённого (Grossularia reclinata (L.) Mill.) сорта «Московский красный» и разработка обоснований для их применения в медицине
- Фукус как лекарственное средство
- Влияние токсичных цветений цианобактерий на организмы зообентоса в Куршском заливе Балтийского моря
- Применение экстрактов клюквы в медицине
- Создание модели экосистемы моря каменноугольного периода Подмосковья
- Сравнительный анализ пыльцевых зёрен у разных видов цветковых растений
- Изучение взаимосвязи между характеристиками лазерных диодов на основе арсенида галлия и областями их применения в медицине
- Медицинский тренажёр-симулятор для обучения мануальным навыкам эндовидеохирургии
- Проблема близорукости у десятиклассников и способы её профилактики
- История диагностики сахарного диабета
- Вакцинопрофилактика гриппа: причины отказов и согласий
- Это удивительное растение – томат, картофель или…?
- Анализ шума в здании школы
- Уникальное вещество – вода. Какую воду мы пьём? Химический анализ воды
- Исследование наследственных признаков голубей при скрещивании разных пород
- Гигиена дыхания
- Лучевая диагностика деформации стоп
- Изменение физико-химического состава слюны человека в зависимости от его психоэмоционального состояния
- Экспертиза качества оливкового масла
- Может ли подросток не лгать?
- Изучение влияния искусственного ультрафиолетового излучения на жизнедеятельность живых организмов
- Исследование влияния правополушарности на развитие школьника
- Анализ лекарственных препаратов из домашней аптечки
- Радиационные и физические методы предотвращения распространения внутрибольничной инфекции
- История развития трансфузиологии
- Исследование кариотипов Simulium vernum species group (Diptera: Simuliidae) в Московской области
- Накопление астаксантина в пищевых цепях и оптимизация его биогенного синтеза
- Зависимость электроактивности бактерий рода Shewanella от рН среды культивирования
- Аптека в холодильнике
- Социальные отношения в группе приматов. Восточный колобус, или гвереца
- Введение в культуру in vitro семян Nepeta mussinii Spreng
- Исследование бактерицидных и кислотно-основных свойств жевательной смолки
- Оценка функционального состояния сердечно-сосудистой системы учащихся
- Микробиологическое изучение воды
- Другая сторона радуги или применение оптической спектроскопии в исследовании состава вещества
- Близорукость у школьников. Причины и профилактика
- Эндофитные дрожжевые группировки в соплодиях ананаса
- Анализ антропометрических показателей физического развития детей 5-7 лет, посещающих дошкольное отделение ГБОУ «Лицей №1586» Западного округа города Москвы
- «Гигиенические требования к условиям обучения в общеобразовательных учреждениях» или Санитарно-эпидемиологические правила и нормативы (СанПиН 2.4.2.1178-02)
- Изучение устойчивости микроорганизмов воздуха школы к различным антибиотикам
- Методы обработки хирургического инструментария
- Модель распространения инфекционных заболеваний
- Флора и краснокнижные виды растений лесов долины реки Серебрянка природно-исторического парка «Измайлово» (город Москва)
- Современный подход к системе контроля знаний в профильных классах
- Изучение методики изготовления препаратов мышечной ткани
- Изучение адсорбирующих материалов для устранения нефтяных разливов в Арктике
- Взаимосвязь между уровнем развития памяти и успешностью обучения учащихся 7−10 классов школы
- Исследование нахождения витаминов в продуктах питания
- Исследование проблемы плоскостопия у школьников
- Исследование качества наиболее часто употребляемых продуктов питания